Ενεργά συστατικά: Εσομεπραζόλη
LUCEN 20 mg γαστροανθεκτικά δισκία
LUCEN 40 mg γαστροανθεκτικά δισκία
Τα ένθετα συσκευασίας Lucen διατίθενται για μεγέθη συσκευασίας: - LUCEN γαστροανθεκτικά δισκία 20 mg, γαστροανθεκτικά δισκία LUCEN 40 mg
- LUCEN 10 mg γαστροανθεκτικά κοκκία για πόσιμο εναιώρημα, σε φακελλίσκο
- LUCEN 40 mg σκόνη για ενέσιμο διάλυμα / έγχυση
Ενδείξεις Γιατί χρησιμοποιείται το Lucen; Σε τι χρησιμεύει;
Το LUCEN περιέχει ένα φάρμακο που ονομάζεται εσομεπραζόλη. Ανήκει σε μια ομάδα φαρμάκων που ονομάζονται «αναστολείς της αντλίας πρωτονίων», τα οποία λειτουργούν μειώνοντας την ποσότητα οξέος που παράγεται από το στομάχι.
Το LUCEN χρησιμοποιείται για τη θεραπεία των ακόλουθων διαταραχών:
- «Γαστροοισοφαγική παλινδρόμηση» (ΓΟΠΝ). Εμφανίζεται όταν το οξύ από το στομάχι διαφεύγει στον οισοφάγο (ο σωλήνας που συνδέει το λαιμό με το στομάχι), προκαλώντας πόνο, φλεγμονή και κάψιμο.
- Έλκη στομάχου ή άνω εντέρου μολυσμένα με βακτήρια που ονομάζονται "Helicobacter pylori". Εάν έχετε αυτές τις καταστάσεις, ο γιατρός σας μπορεί επίσης να συνταγογραφήσει αντιβιοτικά για τη θεραπεία της λοίμωξης και να επιτρέψει στο έλκος να επουλωθεί.
- Έλκη στομάχου που προκαλούνται από φάρμακα που ονομάζονται ΜΣΑΦ (Μη Στεροειδή Αντιφλεγμονώδη Φάρμακα). Το LUCEN μπορεί επίσης να χρησιμοποιηθεί για την πρόληψη του σχηματισμού έλκους στομάχου κατά τη λήψη ΜΣΑΦ.
- Υπερβολικό οξύ στομάχου που προκαλείται από όγκο στο πάγκρεας (σύνδρομο Zollinger-Ellisson).
- Παρατεταμένη θεραπεία της εκ νέου αιμορραγίας των ελκών, μετά από πρόληψη με ενδοφλέβια χορήγηση του Lucen
Αντενδείξεις Όταν το Lucen δεν πρέπει να χρησιμοποιείται
Μην πάρετε το LUCEN:
- σε περίπτωση αλλεργίας (υπερευαισθησίας) στην εσομεπραζόλη ή σε οποιοδήποτε άλλο συστατικό αυτού του φαρμάκου (αναφέρονται στην παράγραφο: Περαιτέρω πληροφορίες).
- εάν είστε αλλεργικοί σε άλλα φάρμακα αναστολέα της αντλίας πρωτονίων (π.χ. παντοπραζόλη, λανσοπραζόλη, ραβεπραζόλη, ομεπραζόλη).
- εάν παίρνετε φάρμακο που περιέχει νελφιναβίρη (χρησιμοποιείται για τη θεραπεία του HIV).
Δεν πρέπει να πάρετε το LUCEN εάν εμπίπτει σε οποιαδήποτε από τις παραπάνω περιπτώσεις. Εάν έχετε αμφιβολίες, συμβουλευτείτε το γιατρό ή το φαρμακοποιό σας πριν πάρετε το LUCEN.
Προφυλάξεις κατά τη χρήση Τι πρέπει να γνωρίζετε πριν πάρετε το Lucen
Προσέξτε ιδιαίτερα με το LUCEN
Μιλήστε με το γιατρό ή το φαρμακοποιό σας πριν πάρετε το LUCEN εάν:
- Έχετε σοβαρά ηπατικά προβλήματα
- Έχετε σοβαρά νεφρικά προβλήματα.
Το LUCEN μπορεί να καλύψει τα συμπτώματα άλλων ασθενειών. Επομένως, εάν σας συμβεί κάποιο από τα παρακάτω πριν αρχίσετε να παίρνετε ή ενώ παίρνετε το LUCEN, ενημερώστε αμέσως το γιατρό σας:
- Χάνετε πολύ βάρος χωρίς λόγο ή δυσκολεύεστε να καταπιείτε
- Εμφανίζεται πόνος στο στομάχι ή δυσπεψία
- Ξεκινήστε να κάνετε εμετό για φαγητό ή αίμα
- Τα κόπρανα είναι μαύρα (αιματοβαμμένα κόπρανα).
Εάν σας έχει συνταγογραφηθεί το LUCEN "ανάλογα με τις ανάγκες", επικοινωνήστε με το γιατρό σας εάν τα συμπτώματα επιμένουν ή αλλάζουν χαρακτηριστικά.
Εάν παίρνετε έναν αναστολέα αντλίας πρωτονίων όπως το LUCEN, ειδικά για περισσότερο από ένα χρόνο, μπορεί να έχετε ελαφρώς αυξημένο κίνδυνο κατάγματος του ισχίου, του καρπού ή της σπονδυλικής στήλης. Εάν έχετε οστεοπόρωση ή παίρνετε κορτικοστεροειδή (που μπορεί να αυξήσουν τον κίνδυνο οστεοπόρωση) συμβουλευτείτε το γιατρό σας
Αλληλεπιδράσεις Ποια φάρμακα ή τρόφιμα μπορεί να αλλάξουν την επίδραση του Lucen
Ενημερώστε το γιατρό ή το φαρμακοποιό σας εάν παίρνετε ή έχετε πάρει πρόσφατα άλλα φάρμακα, συμπεριλαμβανομένων των φαρμάκων που χορηγούνται χωρίς ιατρική συνταγή.
Πράγματι, το LUCEN μπορεί να επηρεάσει τον τρόπο λειτουργίας κάποιων φαρμάκων και ορισμένα φάρμακα μπορεί να επηρεάσουν το LUCEN. Δεν πρέπει να παίρνετε το LUCEN εάν παίρνετε φάρμακο που περιέχει νελφιναβίρη (χρησιμοποιείται για τη θεραπεία του HIV).
Ενημερώστε τον γιατρό ή τον φαρμακοποιό σας εάν παίρνετε οποιοδήποτε από τα ακόλουθα φάρμακα:
- Atazanavir (χρησιμοποιείται για τη θεραπεία του HIV)
- Κλοπιδογρέλη (χρησιμοποιείται για την πρόληψη θρόμβων αίματος)
- Κετοκοναζόλη, ιτρακοναζόλη ή βορικοναζόλη (χρησιμοποιείται για τη θεραπεία λοιμώξεων που προκαλούνται από μύκητες).
- Erlotinib (χρησιμοποιείται για τη θεραπεία του καρκίνου).
- Σιταλοπράμη, ιμιπραμίνη ή κλομιπραμίνη (χρησιμοποιείται για τη θεραπεία της κατάθλιψης).
- Διαζεπάμη (χρησιμοποιείται για τη θεραπεία του άγχους, για χαλάρωση των μυών ή για επιληψία).
- Φαινυτοΐνη (χρησιμοποιείται στην επιληψία) Εάν παίρνετε φαινυτοΐνη, ο γιατρός σας θα πρέπει να σας παρακολουθεί κατά την έναρξη ή τη διακοπή της θεραπείας με LUCEN.
- Φάρμακα που χρησιμοποιούνται για την αραίωση του αίματος, όπως η βαρφαρίνη. Ο γιατρός σας μπορεί να σας παρακολουθεί όταν ξεκινάτε ή σταματάτε τη θεραπεία με LUCEN.
- Κιλοσταζόλη (χρησιμοποιείται για τη θεραπεία της διαλείπουσας χωλότητας - πόνος στα πόδια όταν περπατάτε λόγω ανεπαρκούς παροχής αίματος).
- Σισαπρίδη (χρησιμοποιείται για δυσπεψία και καούρα).
- Διγοξίνη (χρησιμοποιείται για καρδιακά προβλήματα).
- Μεθοτρεξάτη (ένα φάρμακο χημειοθεραπείας που χρησιμοποιείται σε υψηλές δόσεις για τη θεραπεία του καρκίνου) - εάν παίρνετε υψηλές δόσεις μεθοτρεξάτης, ο γιατρός σας μπορεί να διακόψει προσωρινά τη θεραπεία σας με το Lucen.
- Τακρόλιμους (χρησιμοποιείται σε μεταμοσχεύσεις οργάνων)
- Ριφαμπικίνη (χρησιμοποιείται για τη θεραπεία της φυματίωσης).
- Βαλσαμόχορτο (Hypericum perforatum) (χρησιμοποιείται για τη θεραπεία της κατάθλιψης).
Εάν ο γιατρός σας έχει συνταγογραφήσει αντιβιοτικά όπως αμοξικιλλίνη και κλαριθρομυκίνη με LUCEN για τη θεραπεία ελκών που προκαλούνται από λοίμωξη από Helicobacter pylori, είναι πολύ σημαντικό να ενημερώσετε το γιατρό σας για άλλα φάρμακα.
Προειδοποιήσεις Είναι σημαντικό να γνωρίζετε ότι:
Εγκυμοσύνη και θηλασμός
Πριν πάρετε το LUCEN, ενημερώστε το γιατρό σας εάν είστε έγκυος ή θέλετε να μείνετε έγκυος. Ζητήστε τη συμβουλή του γιατρού ή του φαρμακοποιού σας πριν πάρετε οποιοδήποτε φάρμακο. Ο γιατρός σας θα αποφασίσει εάν μπορείτε να πάρετε το LUCEN κατά τη διάρκεια αυτής της περιόδου.
Δεν είναι γνωστό εάν το LUCEN περνά στο μητρικό γάλα, επομένως δεν πρέπει να παίρνετε το LUCEN εάν θηλάζετε.
Λήψη του LUCEN με φαγητό και ποτό
Τα δισκία μπορούν να ληφθούν με πλήρες στομάχι ή με άδειο στομάχι.
Οδήγηση και χειρισμός μηχανών
Το LUCEN είναι απίθανο να επηρεάσει την ικανότητά σας να οδηγείτε ή να χρησιμοποιείτε εργαλεία ή μηχανήματα.
Σημαντικές πληροφορίες σχετικά με ορισμένα συστατικά του LUCEN
Τα γαστροανθεκτικά δισκία LUCEN περιέχουν σακχαρόζη που είναι ένας τύπος ζάχαρης. Εάν σας έχει πει ο γιατρός σας ότι έχετε «δυσανεξία σε ορισμένα σάκχαρα, συμβουλευτείτε τον πριν πάρετε το φάρμακο.
Δοσολογία και τρόπος χρήσης Πώς να χρησιμοποιήσετε το Lucen: Δοσολογία
Πάντοτε να παίρνετε το LUCEN αυστηρά σύμφωνα με τις οδηγίες του γιατρού σας. Εάν έχετε αμφιβολίες, θα πρέπει να συμβουλευτείτε το γιατρό ή το φαρμακοποιό σας.
- Τα γαστροανθεκτικά δισκία LUCEN δεν συνιστώνται για παιδιά ηλικίας κάτω των 12 ετών
- Εάν παίρνετε αυτό το φάρμακο για μεγάλο χρονικό διάστημα, ο γιατρός σας θα σας παρακολουθεί (ιδιαίτερα εάν παίρνετε αυτό το φάρμακο για περισσότερο από ένα χρόνο)
- Εάν ο γιατρός σας έχει πει να πάρετε το φάρμακο όταν χρειάζεται, όπως απαιτείται, ενημερώστε το γιατρό σας εάν αλλάξουν τα συμπτώματά σας.
Λήψη του φαρμάκου
- Μπορείτε να πάρετε τα δισκία οποιαδήποτε στιγμή της ημέρας.
- Μπορείτε να πάρετε τα δισκία με γεμάτο στομάχι ή με άδειο στομάχι.
- Καταπιείτε τα δισκία ολόκληρα με ένα ποτό νερό. Μην μασάτε ή συνθλίβετε τα δισκία καθώς περιέχουν επικαλυμμένους κόκκους που προστατεύουν το φάρμακο από την γαστρική οξύτητα.
Τι να κάνετε εάν αντιμετωπίζετε προβλήματα με την κατάποση των δισκίων
Εάν αντιμετωπίζετε πρόβλημα κατάποσης των δισκίων:
- Βάλτε τα δισκία σε ένα ποτήρι ακίνητο νερό. Δεν πρέπει να χρησιμοποιούνται άλλα υγρά
- Ανακατέψτε μέχρι να διαλυθούν τα δισκία (το μείγμα δεν θα έχει καθαρή εμφάνιση). Πίνετε αμέσως ή τουλάχιστον εντός 30 λεπτών. Τα ανακατεύετε πάντα πριν πιείτε
- Για να βεβαιωθείτε ότι έχετε πάρει όλο το φάρμακο, ξεπλύνετε καλά το ποτήρι γεμίζοντας το μέχρι τη μέση με νερό και ποτό.Τα στερεά σωματίδια περιέχουν το φάρμακο και δεν πρέπει να μασήνονται ή να συνθλίβονται.
Εάν δεν μπορείτε να καταπιείτε, το δισκίο μπορεί να αναμειχθεί με λίγο νερό, να εισαχθεί σε σύριγγα και να χορηγηθεί μέσω ενός σωλήνα απευθείας στο στομάχι (γαστρικός σωλήνας).
Πόση φαρμακευτική αγωγή να πάρετε
- Ο γιατρός σας θα σας συμβουλεύσει για τον αριθμό των δισκίων που πρέπει να πάρετε και για πόσο χρονικό διάστημα. Αυτό είναι συνάρτηση της φυσικής κατάστασης, της ηλικίας και της ηπατικής σας κατάστασης.
- Οι συνήθεις δόσεις δίνονται παρακάτω.
Θεραπεία καούρας που προκαλείται από γαστροοισοφαγική παλινδρόμηση (ΓΟΠΝ):
Ενήλικες και παιδιά από 12 ετών:
- Εάν ο γιατρός σας βρήκε ελαφρά κατεστραμμένο τον οισοφάγο σας, η συνήθης δόση είναι ένα γαστροανθεκτικό δισκίο 40 mg LUCEN μία φορά την ημέρα για 4 εβδομάδες. Ο γιατρός σας μπορεί να σας πει να συνεχίσετε τη θεραπεία, παίρνοντας την ίδια δόση, για άλλες 4 εβδομάδες, σε περίπτωση που ο οισοφάγος δεν έχει επουλωθεί.
- Μετά τη θεραπεία του οισοφάγου, η συνήθης δόση είναι ένα γαστροανθεκτικό δισκίο 20 mg LUCEN μία φορά την ημέρα.
- Εάν ο οισοφάγος δεν έχει υποστεί βλάβη, η συνήθης δόση είναι ένα γαστροανθεκτικό δισκίο LUCEN 20 mg κάθε μέρα. Όταν τα συμπτώματα είναι υπό έλεγχο, ο γιατρός σας θα σας ενημερώσει ότι μπορείτε να πάρετε το φάρμακο όταν χρειάζεται, έως και ένα γαστρεντερικό ανθεκτικό δισκίο. Lucen 20 mg την ημέρα.
- Εάν έχετε σοβαρά ηπατικά προβλήματα, ο γιατρός σας θα σας δώσει χαμηλότερη δόση.
Θεραπεία ελκών που προκαλούνται από λοίμωξη από ελικοβακτηρίδιο του πυλωρού και πρόληψη της επανεμφάνισής τους:
- Ενήλικες από 18 ετών και μετά: η συνήθης δόση είναι ένα γαστροανθεκτικό δισκίο LUCEN 20 mg δύο φορές την ημέρα για μία εβδομάδα.
- Ο γιατρός σας θα σας πει επίσης να πάρετε αντιβιοτικά που ονομάζονται αμοξικιλλίνη και κλαριθρομυκίνη.
Θεραπεία γαστρικών ελκών που προκαλούνται από ΜΣΑΦ (Μη Στεροειδή Αντιφλεγμονώδη Φάρμακα):
- Ενήλικες από 18 ετών και μετά: η συνήθης δόση είναι ένα γαστροανθεκτικό δισκίο LUCEN 20 mg μία φορά την ημέρα για 4 έως 8 εβδομάδες.
Πρόληψη του έλκους στομάχου εάν παίρνετε ΜΣΑΦ (Μη Στεροειδή Αντιφλεγμονώδη Φάρμακα):
- Ενήλικες από 18 ετών και μετά: η συνήθης δόση είναι ένα γαστροανθεκτικό δισκίο LUCEN 20 mg μία φορά την ημέρα.
Θεραπεία της περίσσειας οξέος στομάχου που προκαλείται από ανάπτυξη στο πάγκρεας (σύνδρομο Zollinger-Ellison):
- Ενήλικες από 18 ετών και μετά: η συνήθης δόση είναι ένα γαστροανθεκτικό δισκίο LUCEN 40 mg δύο φορές την ημέρα.
- Ο γιατρός σας θα προσαρμόσει τη δόση ανάλογα με τις ανάγκες σας και θα αποφασίσει επίσης πόσο θα συνεχίσετε τη θεραπεία.
Η μέγιστη δόση είναι 80 mg δύο φορές την ημέρα.
Παρατεταμένη θεραπεία της εκ νέου αιμορραγίας ελκών, μετά από πρόληψη με ενδοφλέβια χορήγηση Lucen:
Η συνήθης δόση είναι ένα δισκίο Lucen 40 mg μία φορά την ημέρα για 4 εβδομάδες.
Υπερδοσολογία Τι πρέπει να κάνετε εάν έχετε πάρει πάρα πολύ Lucen
Εάν πάρετε μεγαλύτερη δόση LUCEN από την κανονική
Εάν έχετε πάρει περισσότερο LUCEN από το συνταγογραφούμενο από το γιατρό σας, ενημερώστε αμέσως το γιατρό ή το φαρμακοποιό σας.
Εάν ξεχάσετε να πάρετε το LUCEN
- Εάν ξεχάσετε να πάρετε μια δόση LUCEN, πάρτε το μόλις το θυμηθείτε. Εάν πλησιάζει η ώρα για την επόμενη δόση σας, παραλείψτε τη χαμένη δόση.
- Μην πάρετε διπλή δόση (δύο δόσεις ταυτόχρονα) για να αναπληρώσετε τη δόση που ξεχάσατε.
Παρενέργειες Ποιες είναι οι παρενέργειες του Lucen
Όπως όλα τα φάρμακα, έτσι και το LUCEN μπορεί να προκαλέσει ανεπιθύμητες ενέργειες, αν και δεν παρουσιάζονται σε όλους τους ανθρώπους.
Εάν παρατηρήσετε κάποια από τις ακόλουθες σοβαρές ανεπιθύμητες ενέργειες, σταματήστε να παίρνετε το LUCEN και επικοινωνήστε αμέσως με το γιατρό σας:
- Ξαφνικός συριγμός, πρήξιμο των χειλιών, της γλώσσας και του λαιμού ή του σώματος, εξάνθημα, λιποθυμία ή δυσκολία στην κατάποση (σοβαρή αλλεργική αντίδραση).
- Ερυθρότητα του δέρματος με φουσκάλες ή ξεφλούδισμα. Σοβαρές φουσκάλες και αιμορραγία μπορεί επίσης να εμφανιστούν στα χείλη, τα μάτια, το στόμα, τη μύτη και τα γεννητικά όργανα. Αυτό μπορεί να είναι "σύνδρομο Stevens-Johnson" ή "τοξική επιδερμική νεκρόλυση".
- Το κίτρινο δέρμα, τα σκούρα ούρα και η κόπωση μπορεί να είναι συμπτώματα ηπατικών προβλημάτων. Αυτές οι επιδράσεις είναι σπάνιες, επηρεάζοντας λιγότερο από 1 στα 1000 άτομα.
Άλλες παρενέργειες περιλαμβάνουν:
Συχνές (επηρεάζουν λιγότερα από 1 στα 10 άτομα):
- Πονοκέφαλο.
- Επιδράσεις στο στομάχι ή στα έντερα: διάρροια, πόνος στο στομάχι, δυσκοιλιότητα, μετεωρισμός.
- Ναυτία ή έμετος.
Όχι συχνές (επηρεάζουν λιγότερα από 1 στα 100 άτομα):
- Πρήξιμο στα πόδια και τους αστραγάλους.
- Διαταραγμένος ύπνος (αϋπνία).
- Ζάλη, καρφίτσες, υπνηλία.
- Ζάλη.
- Ξερό στόμα
- Αλλαγές στις εξετάσεις αίματος που ελέγχουν τον τρόπο λειτουργίας του ήπατος.
- Δερματικό εξάνθημα, κνίδωση και κνησμός.
- Κάταγμα ισχίου, καρπού ή σπονδυλικής στήλης (εάν το Lucen χρησιμοποιείται σε υψηλές δόσεις και για παρατεταμένες περιόδους).
Σπάνια (επηρεάζει λιγότερα από 1 στα 1.000 άτομα):
- Προβλήματα αίματος, όπως μειωμένος αριθμός λευκών αιμοσφαιρίων και αιμοπεταλίων. Αυτό μπορεί να προκαλέσει αδυναμία, μώλωπες ή πιθανότητα ευκολότερης μόλυνσης.
- Χαμηλά επίπεδα νατρίου στο αίμα. Αυτό μπορεί να προκαλέσει αδυναμία, έμετο και κράμπες.
- Αίσθημα διέγερσης, σύγχυσης ή κατάθλιψης.
- Αλλαγές στη γεύση.
- Προβλήματα με την όρασή σας, όπως θολή όραση.
- Ξαφνικός συριγμός ή δύσπνοια (βρογχόσπασμος).
- Φλεγμονή στο εσωτερικό του στόματος.
- Μια λοίμωξη που ονομάζεται «τσίχλα» και μπορεί να επηρεάσει το έντερο και προκαλείται από μύκητα.
- Προβλήματα στο συκώτι, συμπεριλαμβανομένου του ίκτερου που μπορεί να προκαλέσει κίτρινο δέρμα, σκούρα ούρα και κόπωση.
- Τριχόπτωση (αλωπεκία).
- Δερματικό εξάνθημα κατά την έκθεση στον ήλιο.
- Πόνος στις αρθρώσεις (αρθραλγία) ή μυϊκός πόνος (μυαλγία).
- Γενικό αίσθημα αδιαθεσίας και έλλειψη δύναμης.
- Αυξημένη εφίδρωση.
Πολύ σπάνια (επηρεάζει λιγότερα από 1 στα 10.000 άτομα):
- Αλλαγές στον αριθμό των αιμοσφαιρίων, συμπεριλαμβανομένης της ακοκκιοκυτταραιμίας (έλλειψη λευκών αιμοσφαιρίων).
- Επίθεση.
- Βλέποντας, νιώθοντας ή ακούγοντας πράγματα που δεν υπάρχουν (παραισθήσεις).
- Σοβαρά ηπατικά προβλήματα που οδηγούν σε ηπατική ανεπάρκεια και φλεγμονή του εγκεφάλου.
- Ξαφνική εμφάνιση σοβαρού εξανθήματος ή φουσκάλες ή ξεφλούδισμα του δέρματος. Αυτό μπορεί να σχετίζεται με υψηλό πυρετό και πόνο στις αρθρώσεις (πολύμορφο ερύθημα, σύνδρομο Stevens-Johnson, τοξική επιδερμική νεκρόλυση).
- Μυϊκή αδυναμία.
- Σοβαρά νεφρικά προβλήματα.
- Διεύρυνση στήθους στους άνδρες.
Άγνωστο (η συχνότητα δεν μπορεί να εκτιμηθεί από τα διαθέσιμα δεδομένα)
- Εάν πάρετε το LUCEN για περισσότερο από τρεις μήνες, τα επίπεδα μαγνησίου στο αίμα σας μπορεί να μειωθούν. Τα χαμηλά επίπεδα μαγνησίου μπορούν να εκδηλωθούν με κόπωση, ακούσιες μυϊκές συσπάσεις, αποπροσανατολισμό, σπασμούς, ζάλη, αυξημένο καρδιακό ρυθμό. Εάν έχετε οποιοδήποτε από αυτά τα συμπτώματα, συμβουλευτείτε αμέσως το γιατρό σας. Τα χαμηλά επίπεδα μαγνησίου μπορούν επίσης να οδηγήσουν σε μείωση των επιπέδων καλίου ή ασβεστίου στο αίμα. Ο γιατρός σας πρέπει να αποφασίσει εάν θα ελέγχει τα επίπεδα μαγνησίου στο αίμα σας περιοδικά.
- Φλεγμονή στο έντερο (που οδηγεί σε διάρροια).
Το LUCEN μπορεί σε πολύ σπάνιες περιπτώσεις να επηρεάσει τα λευκά αιμοσφαίρια οδηγώντας σε ανοσοανεπάρκεια. Εάν έχετε λοίμωξη με συμπτώματα όπως πυρετό με σοβαρή επιδείνωση της γενικής φυσικής σας κατάστασης ή πυρετό με συμπτώματα τοπικής λοίμωξης, όπως πόνο στο λαιμό, το λαιμό ή το στόμα ή δυσκολία στην ούρηση, θα πρέπει να επισκεφθείτε το γιατρό σας το συντομότερο δυνατόν ότι η έλλειψη λευκών αιμοσφαιρίων (ακοκκιοκυττάρωση) μπορεί να αποκλειστεί μέσω μιας εξέτασης αίματος. Είναι σημαντικό για εσάς να δώσετε πληροφορίες σχετικά με τα φάρμακα που παίρνετε. Μην ανησυχείτε για τη λίστα πιθανών ανεπιθύμητων ενεργειών παραπάνω. Ενδέχεται να μην εμφανίσετε καμία. Εάν κάποια ανεπιθύμητη ενέργεια γίνει σοβαρή ή αν παρατηρήσετε κάποια ανεπιθύμητη ενέργεια που δεν αναφέρεται στο παρόν φύλλο οδηγιών, ενημερώστε το γιατρό ή το φαρμακοποιό σας.
Λήξη και διατήρηση
- Να φυλάσσεται σε μέρη που δεν το φθάνουν και δεν το βλέπουν τα παιδιά.
- Μη φυλάσσετε σε θερμοκρασία μεγαλύτερη των 30 ° C.
- Φυλάσσετε στην αρχική συσκευασία (blister) ή διατηρείτε το δοχείο ερμητικά κλειστό (μπουκάλι) για να προστατεύεται από την υγρασία.
- Μη χρησιμοποιείτε τα δισκία μετά την ημερομηνία λήξης (ΛΗΞΗ) που αναφέρεται στο κουτί, στο πορτοφόλι ή στην κυψέλη. Η ημερομηνία λήξης αναφέρεται στην τελευταία ημέρα του μήνα.
- Τα φάρμακα δεν πρέπει να απορρίπτονται στα λύματα ή στα οικιακά απορρίμματα. Ρωτήστε τον φαρμακοποιό σας πώς να πετάξετε τα φάρμακα που δεν χρησιμοποιείτε πια. Αυτό θα βοηθήσει στην προστασία του περιβάλλοντος.
ΑΛΛΕΣ ΠΛΗΡΟΦΟΡΙΕΣ
Τι περιέχει το LUCEN
Το δραστικό συστατικό είναι η εσομεπραζόλη. Τα γαστροανθεκτικά δισκία LUCEN υπάρχουν σε 2 περιεκτικότητες που περιέχουν 20 ή 40 mg εσομεπραζόλης (ως τριένυδρο μαγνήσιο).
Τα άλλα συστατικά είναι: μονοστεατική γλυκερόλη 40-55, υπολόζη, υπρομελλόζη, οξείδιο του σιδήρου (κόκκινο-καφέ, κίτρινο) (Ε172, μόνο για δισκία 20 mg), στεατικό μαγνήσιο, συμπολυμερές μεθακρυλικού οξέος αιθυλ ακρυλικός εστέρας (1: 1) στα 30 %, μικροκρυσταλλική κυτταρίνη, συνθετική παραφίνη, μακρογκόλες, πολυσορβικό 80, κροσποβιδόνη, στεατυλο φουμαρικό νάτριο, σφαίρες σακχαρόζης (σακχαρόζη και άμυλο καλαμποκιού), τάλκης, διοξείδιο του τιτανίου (Ε171), κιτρικό τριαιθύλιο.
Περιγραφή της εμφάνισης του LUCEN και του περιεχομένου της συσκευασίας
- Τα γαστροανθεκτικά δισκία LUCEN 20 mg είναι ανοιχτό ροζ με A / EH στη μία πλευρά και 20 mg στην άλλη.
- Τα γαστροανθεκτικά δισκία LUCEN 40 mg είναι ροζ με A / EI στη μία πλευρά και 40 mg στην άλλη.
- Τα δισκία βρίσκονται σε συσκευασίες κυψέλης, πορτοφόλια και / ή μπουκάλια που περιέχουν
- 20 mg, 40 mg: μπουκάλι 2-5-7-14-15-28-30-56-60-100-140 (28x5) δισκία.
- 20 mg, 40 mg: blister ή πορτοφόλι blister από 3-7-7x1-14-15-25x1-28-30-50x1- 56-60-90-98-100x1-140 δισκία.
Μπορεί να μην κυκλοφορούν όλες οι συσκευασίες
Φύλλο οδηγιών χρήσης: AIFA (Ιταλικός Οργανισμός Φαρμάκων). Περιεχόμενο που δημοσιεύτηκε τον Ιανουάριο του 2016. Οι πληροφορίες που υπάρχουν δεν μπορεί να είναι ενημερωμένες.
Για να έχετε πρόσβαση στην πιο ενημερωμένη έκδοση, είναι σκόπιμο να αποκτήσετε πρόσβαση στον ιστότοπο AIFA (Ιταλικός Οργανισμός Φαρμάκων). Αποποίηση ευθυνών και χρήσιμες πληροφορίες.
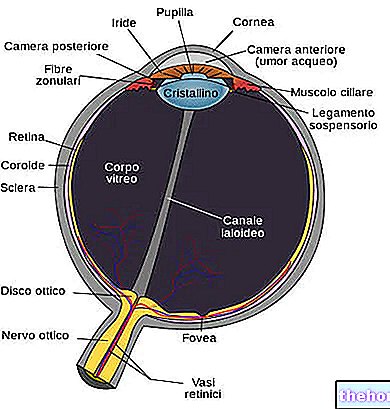
01.0 ΟΝΟΜΑ ΤΟΥ ΦΑΡΜΑΚΕΥΤΙΚΟΥ ΠΡΟDΟΝΤΟΣ
LUCENTIS 10 MG / ML ΔΙΑΛΥΜΑ ΓΙΑ ΕΝΕΣΗ
02.0 ΠΟΙΟΤΙΚΗ ΚΑΙ ΠΟΣΟΤΙΚΗ ΣΥΝΘΕΣΗ
Ένα ml περιέχει 10 mg ranibizumab *. Κάθε φιαλίδιο περιέχει 2,3 mg ranibizumab σε 0,23 ml διαλύματος.
* Το Ranibizumab είναι ένα εξανθρωπισμένο μονοκλωνικό θραύσμα αντισώματος που παράγεται στα κύτταρα του Escherichia coli με τεχνολογία ανασυνδυασμένου DNA.
Για τον πλήρη κατάλογο των εκδόχων, ανατρέξτε στην ενότητα 6.1.
03.0 ΦΑΡΜΑΚΟΤΕΧΝΙΚΗ ΜΟΡΦΗ
Ενέσιμο διάλυμα
Διαυγές, άχρωμο έως ωχροκίτρινο υδατικό διάλυμα.
04.0 ΚΛΙΝΙΚΕΣ ΠΛΗΡΟΦΟΡΙΕΣ
04.1 Θεραπευτικές ενδείξεις
Το Lucentis ενδείκνυται σε ενήλικες για:
• Θεραπεία νεοαγγειακής (υγρής) εκφύλισης της ωχράς κηλίδας (AMD)
• Θεραπεία της όρασης που προκαλείται από διαβητικό οίδημα της ωχράς κηλίδας (DME)
• Θεραπεία της οπτικής δυσλειτουργίας που προκαλείται από «οίδημα της ωχράς κηλίδας δευτερογενή σε απόφραξη της φλέβας του αμφιβληστροειδούς (RVO κλάδου ή κεντρικό RVO)
• Θεραπεία της οπτικής δυσλειτουργίας που προκαλείται από χοριοειδή νεοαγγείωση (CNV) δευτερογενώς από παθολογική μυωπία (PM)
04.2 Δοσολογία και τρόπος χορήγησης
Το Lucentis πρέπει να χορηγείται από εξειδικευμένο οφθαλμίατρο με εμπειρία σε ενδοϋαλοειδείς ενέσεις.
Δοσολογία για τη θεραπεία υγρού AMD
Η συνιστώμενη δόση του Lucentis είναι 0,5 mg που χορηγείται μηνιαίως ως εφάπαξ ενδοϋαλοειδής ένεση. Αυτό αντιστοιχεί σε έναν ενέσιμο όγκο 0,05 ml.
Η θεραπεία χορηγείται μηνιαίως και συνεχίζεται έως ότου επιτευχθεί η μέγιστη οπτική οξύτητα, δηλαδή η οπτική οξύτητα του ασθενούς είναι σταθερή για τρεις συνεχόμενους μηνιαίους ελέγχους που πραγματοποιούνται κατά τη διάρκεια της θεραπείας με ranibizumab.
Επομένως, η οπτική οξύτητα των ασθενών πρέπει να παρακολουθείται μηνιαίως.
Η θεραπεία θα πρέπει να ξαναρχίσει όταν η παρακολούθηση υποδεικνύει μείωση της οπτικής οξύτητας λόγω υγρού AMD. Στη συνέχεια θα πρέπει να χορηγούνται μηνιαίες ενέσεις έως ότου επιτευχθεί ξανά σταθερή οπτική οξύτητα για τρεις διαδοχικούς μηνιαίους ελέγχους (αυτό συνεπάγεται τουλάχιστον δύο ενέσεις). Το διάστημα μεταξύ δύο δόσεων δεν πρέπει να είναι μικρότερο από ένα μήνα.
Δοσολογία για τη θεραπεία της οπτικής δυσλειτουργίας που προκαλείται από DME ή οίδημα της ωχράς κηλίδας δευτερογενή σε RVO
Η συνιστώμενη δόση του Lucentis είναι 0,5 mg που χορηγείται μηνιαίως ως εφάπαξ ενδοϋαλοειδής ένεση. Αυτό αντιστοιχεί σε έναν ενέσιμο όγκο 0,05 ml.
Η θεραπεία χορηγείται μηνιαίως και συνεχίζεται έως ότου επιτευχθεί η μέγιστη οπτική οξύτητα, δηλαδή η οπτική οξύτητα του ασθενούς είναι σταθερή για τρεις συνεχόμενους μηνιαίους ελέγχους που πραγματοποιούνται κατά τη διάρκεια της θεραπείας με ranibizumab. Εάν δεν υπάρχει βελτίωση στην οπτική οξύτητα κατά την περίοδο των τριών πρώτων ενέσεων, δεν συνιστάται η συνέχιση της θεραπείας.
Επομένως, η οπτική οξύτητα των ασθενών πρέπει να παρακολουθείται μηνιαίως.
Η θεραπεία θα πρέπει να ξαναρχίσει όταν η παρακολούθηση υποδεικνύει μειωμένη οπτική οξύτητα λόγω DME ή οίδημα της ωχράς κηλίδας δευτερογενή σε RVO. Στη συνέχεια, θα πρέπει να χορηγούνται μηνιαίες ενέσεις έως ότου επιτευχθεί ξανά σταθερή οπτική οξύτητα για τρεις μηνιαίους ελέγχους. Διαδοχικά (αυτό περιλαμβάνει τουλάχιστον δύο ενέσεις). Το διάστημα μεταξύ των δύο δόσεων δεν πρέπει να είναι μικρότερο από ένα μήνα.
Lucentis και φωτοπηξία με λέιζερ σε DME και οίδημα της ωχράς κηλίδας δευτερογενή σε BRVO
Υπάρχει κάποια εμπειρία από τη χορήγηση του Lucentis ταυτόχρονα με φωτοπηξία με λέιζερ (βλέπε παράγραφο 5.1).Όταν χορηγείται την ίδια ημέρα, το Lucentis πρέπει να χορηγείται τουλάχιστον 30 λεπτά μετά τη φωτοπηξία με λέιζερ. Το Lucentis μπορεί να χορηγηθεί σε ασθενείς που έχουν λάβει προηγουμένως φωτοπηξία με λέιζερ.
Δοσολογία για τη θεραπεία της οπτικής δυσλειτουργίας που προκαλείται από το CNV δευτερογενώς σε PM
Η θεραπεία πρέπει να ξεκινά με μία μόνο ένεση.
Εάν η παρακολούθηση αποκαλύψει σημάδια δραστηριότητας της νόσου, όπως μειωμένη οπτική οξύτητα και / ή σημεία τραυματισμού, συνιστάται περαιτέρω θεραπεία.
Η παρακολούθηση των ασθενειών μπορεί να περιλαμβάνει κλινική εξέταση, τομογραφία οπτικής συνοχής (OCT) ή αγγειογραφία φθορισμοειδών (FA).
Ενώ ορισμένοι ασθενείς μπορεί να χρειαστούν μόνο μία ή δύο ενέσεις κατά τη διάρκεια του πρώτου έτους θεραπείας, μερικοί μπορεί να χρειάζονται πιο συχνή θεραπεία (βλ. Παράγραφο 5.1). Συνεπώς, συνιστάται μηνιαία παρακολούθηση για τους πρώτους δύο μήνες και τουλάχιστον κάθε τρεις μήνες κατά τη διάρκεια του πρώτου έτους θεραπείας. Μετά το πρώτο έτος, η συχνότητα της παρακολούθησης μπορεί να καθοριστεί από το γιατρό.
Το διάστημα μεταξύ δύο δόσεων δεν πρέπει να είναι μικρότερο από ένα μήνα.
Lucentis και φωτοδυναμική θεραπεία με Visudyne σε CNV δευτερεύουσα σε PM
Δεν υπάρχει εμπειρία με τη χορήγηση του Lucentis σε συνδυασμό με το Visudyne.
Ειδικοί πληθυσμοί
Ηπατική ανεπάρκεια
Το Lucentis δεν έχει μελετηθεί σε ασθενείς με ηπατική ανεπάρκεια. Ωστόσο, δεν απαιτούνται ιδιαίτερες εκτιμήσεις για αυτήν την πόλωση.
Νεφρική ανεπάρκεια
Δεν απαιτείται προσαρμογή της δόσης σε ασθενείς με νεφρική ανεπάρκεια (βλ. Παράγραφο 5.2).
Ατομα της τρίτης ηλικίας
Δεν απαιτείται προσαρμογή της δόσης στους ηλικιωμένους. Υπάρχει "περιορισμένη" εμπειρία σε ασθενείς με DME άνω των 75 ετών.
Παιδιατρικός πληθυσμός
Η ασφάλεια και η αποτελεσματικότητα του Lucentis σε παιδιά και εφήβους κάτω των 18 ετών δεν έχουν τεκμηριωθεί. Δεν υπάρχουν διαθέσιμα δεδομένα.
Τρόπος χορήγησης
Φιαλίδια μίας χρήσης μόνο για ενδοϋαλοειδή χρήση.
Πριν από τη χορήγηση, το Lucentis πρέπει να ελέγχεται οπτικά για την παρουσία σωματιδίων και αποχρωματισμού.
Η διαδικασία για την ένεση πρέπει να εκτελείται υπό άσηπτες συνθήκες, οι οποίες περιλαμβάνουν απολύμανση των χεριών όπως για κάθε χειρουργική επέμβαση, αποστειρωμένα γάντια, αποστειρωμένη κουρτίνα και αποστειρωμένο βλεφαροστάτη (ή ισοδύναμο) και δυνατότητα διενέργειας στείρας παρακέντησης (εάν είναι απαραίτητο το ιστορικό αντιδράσεων υπερευαισθησίας του ασθενούς πρέπει να αξιολογηθεί προσεκτικά πριν από την ενδοϋαλοειδή διαδικασία (βλ. παράγραφο 4.4). Πρέπει να χορηγείται επαρκής αναισθησία και τοπικό αντιμικροβιακό ευρέως φάσματος πριν από την ένεση για την απολύμανση της περιφθαλμικής, οφθαλμικής και βλεφάρικης επιφάνειας, σύμφωνα με την κλινική πρακτική.
Για πληροφορίες σχετικά με την παρασκευή του Lucentis, ανατρέξτε στην ενότητα 6.6.
Εισάγετε τη βελόνα ένεσης 3,5-4,0 mm πίσω από τον άκρο, στον υαλοειδή θάλαμο, αποφεύγοντας τον οριζόντιο μεσημβρινό και κατευθύνοντας τη βελόνα προς το κέντρο του βολβού του ματιού. Εγχέστε τον όγκο της ένεσης 0,05 ml · αλλάξτε τη θέση του σκληρού χιτώνα για τις επόμενες ενέσεις.
04.3 Αντενδείξεις
Υπερευαισθησία στη δραστική ουσία ή σε κάποιο από τα έκδοχα που αναφέρονται στην παράγραφο 6.1.
Ασθενείς με τρέχουσες ή υπόνοιες οφθαλμικές ή περιφθαλμικές λοιμώξεις.
Ασθενείς με συνεχή σοβαρή ενδοφθάλμια φλεγμονή.
04.4 Ειδικές προειδοποιήσεις και κατάλληλες προφυλάξεις κατά τη χρήση
Αντιδράσεις που σχετίζονται με ενδοϋαλοειδή ένεση
Ενδοϋαλοειδείς ενέσεις, συμπεριλαμβανομένων αυτών με Lucentis, έχουν συσχετιστεί με ενδοφθαλμίτιδα, ενδοφθάλμια φλεγμονή, ρεγματογενή αποκόλληση αμφιβληστροειδούς, ρήξη αμφιβληστροειδούς και ιατρογενή τραυματικό καταρράκτη (βλ. Παράγραφο 4.8). Για τη χορήγηση του Lucentis πρέπει πάντα να χρησιμοποιούνται κατάλληλες τεχνικές ασηπτικής ένεσης. Επιπλέον, οι ασθενείς θα πρέπει να παρακολουθούνται την εβδομάδα μετά την ένεση για να επιτρέπεται η ταχεία θεραπεία σε περίπτωση λοίμωξης. Οι ασθενείς θα πρέπει να ενημερώνονται για το πώς να αναφέρουν χωρίς καθυστέρηση τυχόν συμπτώματα που υποδηλώνουν ενδοφθαλμίτιδα ή οποιοδήποτε από τα παραπάνω συμβάντα.
Αυξήσεις της ενδοφθάλμιας πίεσης
Παρατηρήθηκαν παροδικές αυξήσεις της ενδοφθάλμιας πίεσης (IOP) εντός 60 λεπτών από την ένεση του Lucentis. Παρατηρήθηκαν επίσης παρατεταμένες αυξήσεις της IOP (βλ. Παράγραφο 4.8). Η ενδοφθάλμια πίεση και η αιμάτωση του οπτικού νεύρου πρέπει να παρακολουθούνται και να αντιμετωπίζονται κατάλληλα.
Διμερής θεραπεία
Περιορισμένα δεδομένα για τη διμερή χρήση του Lucentis (συμπεριλαμβανομένης της δοσολογίας την ίδια ημέρα) δεν υποδεικνύουν αυξημένο κίνδυνο συστηματικών ανεπιθύμητων ενεργειών σε σύγκριση με τη μονομερή θεραπεία.
Ανοσογονικότητα
Υπάρχει πιθανότητα ανοσογονικότητας με το Lucentis. Δεδομένου ότι υπάρχει πιθανότητα αυξημένης συστηματικής έκθεσης σε άτομα με DME, δεν μπορεί να αποκλειστεί ο αυξημένος κίνδυνος ανάπτυξης υπερευαισθησίας σε αυτόν τον πληθυσμό ασθενών. Οι ασθενείς θα πρέπει επίσης να εκπαιδεύονται για το πώς να αναφέρουν εάν η ενδοφθάλμια φλεγμονή επιδεινωθεί, επειδή θα μπορούσε να είναι ένα κλινικό σύμπτωμα που οφείλεται σε ο σχηματισμός ενδοφθάλμιων αντισωμάτων.
Ταυτόχρονη χρήση με άλλα αντι-VEGF (αγγειακός ενδοθηλιακός αυξητικός παράγοντας)
Το Lucentis δεν πρέπει να χορηγείται ταυτόχρονα με άλλα φαρμακευτικά προϊόντα αντι-VEGF (συστηματικά ή οφθαλμικά).
Διακοπή Lucentis
Η δόση δεν πρέπει να χορηγείται και η θεραπεία δεν πρέπει να επαναλαμβάνεται πριν από την επόμενη προγραμματισμένη θεραπεία στην περίπτωση:
• μείωση της καλύτερης διορθωμένης οπτικής οξύτητας (BCVA) letters 30 γράμματα σε σύγκριση με την τελευταία αξιολόγηση.
• ενδοφθάλμια πίεση ≥30 mmHg.
• ρήξη αμφιβληστροειδούς.
• «υποεμφιβληστροειδής αιμορραγία που εκτείνεται στο κέντρο του όρμου ή εάν η έκταση της αιμορραγίας είναι ≥50% της συνολικής περιοχής βλάβης».
• ενδοφθάλμια χειρουργική επέμβαση που πραγματοποιήθηκε ή σχεδιάστηκε εντός των προηγούμενων ή επόμενων 28 ημερών.
Ρήξη του επιθηλίου χρωστικής του αμφιβληστροειδούς
Οι παράγοντες κινδύνου που σχετίζονται με την έναρξη ρήξης επιθηλιακής χρωστικής αμφιβληστροειδούς μετά από θεραπεία με αντι-VEGF για υγρή AMD περιλαμβάνουν μεγάλη ή / και υψηλή αποκόλληση επιθηλιακής χρωστικής αμφιβληστροειδούς. Κατά την έναρξη της θεραπείας με Lucentis, θα πρέπει να δίνεται προσοχή σε ασθενείς με αυτούς τους παράγοντες κινδύνου για ρήξη του επιθηλίου χρωστικής του αμφιβληστροειδούς.
Ραγματογενής αποκόλληση αμφιβληστροειδούς ή οπές ωχράς κηλίδας
Η θεραπεία πρέπει να διακόπτεται σε άτομα με ρεγματογενή αποκόλληση αμφιβληστροειδούς ή στάδια 3 ή 4 οπών της ωχράς κηλίδας.
Πληθυσμοί με περιορισμένα δεδομένα
Υπάρχει μόνο περιορισμένη εμπειρία στη θεραπεία ατόμων με DME δευτεροπαθούς διαβήτη τύπου Ι. Το Lucentis δεν έχει μελετηθεί σε ασθενείς που είχαν λάβει προηγουμένως ενδοϋαλοειδείς ενέσεις, σε ασθενείς με ενεργές συστηματικές λοιμώξεις, πολλαπλασιαστική διαβητική αμφιβληστροειδοπάθεια ή σε ασθενείς με ταυτόχρονη ιατρική πάθηση Δεν υπάρχει επίσης εμπειρία από τη θεραπεία με Lucentis σε διαβητικούς ασθενείς με HbAlc μεγαλύτερο από 12% και ανεξέλεγκτη υπέρταση. Η έλλειψη πληροφοριών θα πρέπει να λαμβάνεται υπόψη από τον ιατρό κατά τη θεραπεία αυτών των ασθενών.
Σε ασθενείς με PM, υπάρχουν περιορισμένα δεδομένα για την επίδραση του Lucentis σε ασθενείς που προηγουμένως έλαβαν θεραπεία με αποτυχημένη φωτοδυναμική θεραπεία με βερτεπορφίνη (vPDT). Επιπλέον, ενώ παρατηρήθηκε συνεπής επίδραση σε άτομα με υπο -σπονδυλικές και πλευρικές βλάβες, δεν υπάρχουν επαρκή δεδομένα για την επίδραση του Lucentis σε άτομα με PM με εξωστοματικές βλάβες.
Συστηματικές επιδράσεις μετά από ενδοϋαλοειδή χορήγηση
Συστηματικά ανεπιθύμητα συμβάντα, συμπεριλαμβανομένων μη οφθαλμικών αιμορραγιών και αρτηριακών θρομβοεμβολικών συμβάντων, έχουν αναφερθεί μετά από ενδοϋαλοειδή ένεση αναστολέων VEGF.
Υπάρχουν περιορισμένα δεδομένα σχετικά με την ασφάλεια της θεραπείας του DME, οίδημα της ωχράς κηλίδας που προκαλείται από RVO και CNV δευτερογενώς σε PM σε ασθενείς με ιστορικό εγκεφαλικού επεισοδίου ή παροδικών ισχαιμικών κρίσεων. Ιδιαίτερη προσοχή πρέπει να δίνεται κατά τη θεραπεία τέτοιων ασθενών (βλ. Παράγραφο 4.8).
Προηγούμενα επεισόδια RVO, ισχαιμικού κλάδου και κεντρικού RVO
Υπάρχει περιορισμένη εμπειρία στη θεραπεία ασθενών με προηγούμενα επεισόδια RVO και ασθενών με ισχαιμικό κλάδο RVO (BRVO) και κεντρικό RVO (CRVO). Σε ασθενείς με RVO που παρουσιάζουν απώλεια της οπτικής λειτουργίας με κλινικά σημεία μη αναστρέψιμης ισχαιμίας, η θεραπεία είναι δεν συνιστάται.
04.5 Αλληλεπιδράσεις με άλλα φαρμακευτικά προϊόντα και άλλες μορφές αλληλεπίδρασης
Δεν έχουν διεξαχθεί συμβατικές μελέτες αλληλεπίδρασης.
Για τη συνδυασμένη χρήση φωτοδυναμικής θεραπείας (PDT) με βερτεπορφίνη και Lucentis σε υγρό AMD και PM, βλέπε παράγραφο 5.1.
Για τη συνδυασμένη χρήση φωτοπηξίας λέιζερ και Lucentis στη θεραπεία DME και BRVO, βλέπε παραγράφους 4.2 και 5.1.
04.6 Κύηση και γαλουχία
Γυναίκες σε αναπαραγωγική ηλικία / αντισύλληψη σε γυναίκες
Οι γυναίκες σε αναπαραγωγική ηλικία πρέπει να χρησιμοποιούν αποτελεσματική αντισύλληψη κατά τη διάρκεια της θεραπείας.
Εγκυμοσύνη
Για τη ρανιμπιζουμάμπη, δεν είναι διαθέσιμα κλινικά δεδομένα για εκτεθειμένες εγκυμοσύνες.Μελέτες σε πιθήκους cynomolgus δεν έδειξαν άμεσες ή έμμεσες επιβλαβείς επιδράσεις σε σχέση με την εγκυμοσύνη ή την εμβρυϊκή / εμβρυϊκή ανάπτυξη (βλ. Παράγραφο 5.3). Η συστηματική έκθεση στη ρανιμπιζουμάμπη είναι χαμηλή μετά από οφθαλμική χορήγηση, αλλά λόγω του μηχανισμού δράσης η ρανιμπιζουμάμπη θα πρέπει να θεωρείται δυνητικά τερατογόνος και έμβρυο / εμβρυοτοξική. Επομένως, το ranibizumab δεν πρέπει να χρησιμοποιείται κατά τη διάρκεια της εγκυμοσύνης, εκτός εάν τα αναμενόμενα οφέλη υπερτερούν του δυνητικού κινδύνου για το έμβρυο. Οι γυναίκες που σχεδιάζουν να μείνουν έγκυες και έχουν λάβει θεραπεία με ranibizumab συνιστώνται να περιμένουν τουλάχιστον 3 μήνες μετά την τελευταία δόση ranibizumab πριν συλλάβουν ένα μωρό.
Εγκυμοσύνη
Δεν είναι γνωστό εάν το Lucentis απεκκρίνεται στο ανθρώπινο γάλα. Συνιστάται να μη θηλάζετε κατά τη χρήση του Lucentis.
Γονιμότητα
Δεν υπάρχουν διαθέσιμα στοιχεία για τη γονιμότητα.
04.7 Επιδράσεις στην ικανότητα οδήγησης και χειρισμού μηχανών
Η διαδικασία θεραπείας με Lucentis μπορεί να προκαλέσει παροδικές διαταραχές της όρασης που μπορεί να επηρεάσουν την ικανότητα οδήγησης ή χειρισμού μηχανών (βλ. Παράγραφο 4.8). Οι ασθενείς που εμφανίζουν αυτά τα συμπτώματα δεν πρέπει να οδηγούν ή να χειρίζονται μηχανήματα μέχρι να σταματήσουν αυτές οι παροδικές οπτικές διαταραχές.
04.8 Ανεπιθύμητες ενέργειες
Περίληψη του προφίλ ασφαλείας
Οι περισσότερες ανεπιθύμητες ενέργειες που αναφέρθηκαν μετά τη χορήγηση του Lucentis σχετίζονται με τη διαδικασία ενδοϋαλοειδούς ένεσης.
Οι πιο συχνά αναφερόμενες οφθαλμικές ανεπιθύμητες ενέργειες μετά την ένεση Lucentis είναι: πόνος στα μάτια, υπεραιμία του οφθαλμού, αυξημένη ενδοφθάλμια πίεση, υαλοειδίτιδα, αποκόλληση υαλοειδούς, αιμορραγία αμφιβληστροειδούς, διαταραχή της όρασης, επιπλεύσεις (υαλοειδείς πλωτήρες), αιμορραγία επιπεφυκότα, ερεθισμός των ματιών, αίσθηση ξένου σώματος μάτι, αυξημένο δάκρυ, βλεφαρίτιδα, ξηροφθαλμία και φαγούρα στα μάτια.
Οι πιο συχνά αναφερόμενες μη οφθαλμικές ανεπιθύμητες ενέργειες είναι ο πονοκέφαλος, η ρινοφαρυγγίτιδα και η αρθραλγία.
Λιγότερο συχνά αναφερόμενες αλλά πιο σοβαρές ανεπιθύμητες ενέργειες περιλαμβάνουν ενδοφθαλμίτιδα, τύφλωση, αποκόλληση αμφιβληστροειδούς, ρήξη αμφιβληστροειδούς και ιατρογενή τραυματικό καταρράκτη (βλ. Παράγραφο 4.4).
Οι ασθενείς θα πρέπει να ενημερώνονται για τα συμπτώματα αυτών των πιθανών ανεπιθύμητων ενεργειών και να ενημερώνονται για τον γιατρό τους εάν εμφανίσουν σημεία όπως πόνο στα μάτια ή αυξημένη ενόχληση, επιδείνωση της ερυθρότητας των ματιών, θολή ή μειωμένη όραση, αυξημένο αριθμό υαλοειδών πλωτήρων ή αυξημένη ευαισθησία στο φως.
Οι ανεπιθύμητες ενέργειες που αναφέρθηκαν μετά από χορήγηση του Lucentis σε κλινικές μελέτες συνοψίζονται στον παρακάτω πίνακα.
Πίνακας ανεπιθύμητων αντιδράσεων #
Οι ανεπιθύμητες ενέργειες αναφέρονται κατά κατηγορία οργάνου συστήματος και συχνότητα χρησιμοποιώντας την ακόλουθη σύμβαση: πολύ συχνές (/1 / 10), συχνές (≥1 / 100,
Λοιμώξεις και προσβολές
Πολύ κοινό Ρινοφαρυγγίτιδα
κοινός Λοίμωξη του ουροποιητικού συστήματος *
Διαταραχές του αίματος και του λεμφικού συστήματος
κοινός Αναιμία
Διαταραχές του ανοσοποιητικού συστήματος
κοινός Υπερευαισθησία
Ψυχιατρικές διαταραχές
κοινός Ανησυχία
Διαταραχές του νευρικού συστήματος
Πολύ κοινό Πονοκέφαλο
Διαταραχές των ματιών
Πολύ κοινό Υαλοειδίτιδα, αποκόλληση υαλοειδούς, αιμορραγία αμφιβληστροειδούς, διαταραχές της όρασης, πόνος στα μάτια, υαλοειδείς αιμορραγίες, επιπεφυκότα, ερεθισμός των ματιών, αίσθηση ξένου σώματος στο μάτι, αυξημένη δακρύρροια, βλεφαρίτιδα, ξηροφθαλμία, υπεραιμία του οφθαλμού, φαγούρα στα μάτια.
κοινός Εκφύλιση του αμφιβληστροειδούς, διαταραχές του αμφιβληστροειδούς, αποκόλληση του αμφιβληστροειδούς, αποκόλληση του αμφιβληστροειδικού χρωστικού επιθηλίου, ρήξη του επιθηλίου χρωστικής του αμφιβληστροειδούς, μειωμένη οπτική οξύτητα, αιμορραγία υαλοειδούς, υαλοειδείς διαταραχές, ραγοειδίτιδα, ιρίτιδα, ιριδοκυκλίτιδα, καταρράκτης, υποκάψια, καταρράκτης κερατίτιδα, τριβή κερατοειδούς, αντίδραση πρόσθιου θαλάμου, θολή όραση, αιμορραγία στο σημείο της ένεσης, αιμορραγία στα μάτια, επιπεφυκίτιδα, επιπεφυκίτιδα
αλλεργική, οφθαλμική έκκριση, φωτεινές αναλαμπές, φωτοφοβία, οφθαλμική δυσφορία, οίδημα βλεφάρων, πόνος στα βλέφαρα, υπεραιμία επιπεφυκότα.
Ασυνήθης Τυφλότητα, ενδοφθαλμίτιδα, υποπίπιο, υπόθεμα, κερατοπάθεια, σύννεφα ίριδας, εναποθέσεις κερατοειδούς, οίδημα κερατοειδούς, ραβδώσεις κερατοειδούς, πόνος στο σημείο της ένεσης, ερεθισμός στο σημείο της ένεσης, ανώμαλη αίσθηση στο μάτι, ερεθισμός των βλεφάρων.
Διαταραχές του αναπνευστικού, του θώρακα και του μεσοθωρακίου
κοινός Βήχας
Γαστρεντερικές διαταραχές
κοινός Ναυτία
Διαταραχές του δέρματος και του υποδόριου ιστού
κοινός Αλλεργικές αντιδράσεις (εξάνθημα, κνίδωση, κνησμός, ερύθημα)
Διαταραχές του μυοσκελετικού και του συνδετικού ιστού
Πολύ κοινό Αρθραλγία
Διαγνωστικές εξετάσεις
Πολύ κοινό Αυξημένη ενδοφθάλμια πίεση
# Οι ανεπιθύμητες ενέργειες ορίστηκαν ως ανεπιθύμητες ενέργειες (σε τουλάχιστον 0,5 ποσοστιαίες μονάδες ασθενών) που εμφανίστηκαν σε υψηλότερο ποσοστό (τουλάχιστον 2 ποσοστιαίες μονάδες) σε ασθενείς που λάμβαναν θεραπεία με Lucentis 0,5 mg σε σύγκριση με εκείνους που έλαβαν θεραπεία ελέγχου (απάτη ή PDT βερτεπορφίνη).
* παρατηρείται μόνο στον πληθυσμό με DME
Ανεπιθύμητες ενέργειες που σχετίζονται με την κατηγορία των φαρμάκων
Στη φάση III των υγρών μελετών AMD, η συνολική συχνότητα μη οφθαλμικών αιμορραγιών, ένα ανεπιθύμητο συμβάν που πιθανώς σχετίζεται με τους αναστολείς του VEGF (αυξητικός παράγοντας των ενδοθηλιακών αγγείων), αυξήθηκε ελαφρώς σε ασθενείς που έλαβαν θεραπεία με ranibizumab. Ωστόσο, δεν υπήρξε μοτίβο μεταξύ των διαφορετικών αιμορραγιών. Υπάρχει θεωρητικός κίνδυνος αρτηριακών θρομβοεμβολικών επεισοδίων, συμπεριλαμβανομένου εγκεφαλικού επεισοδίου και εμφράγματος του μυοκαρδίου, που προκύπτει από την ενδοϋαλοειδή χρήση αναστολέων VEGF. Χαμηλή επίπτωση αρτηριακών θρομβοεμβολικών συμβάντων παρατηρήθηκε σε κλινικές δοκιμές με το Lucentis σε ασθενείς με AMD, DME, RVO και PM και δεν παρατηρήθηκαν διαφορές μεταξύ των ομάδων ranibizumab σε σύγκριση με τον έλεγχο.
Αναφορά ύποπτων ανεπιθύμητων ενεργειών
Η αναφορά ύποπτων ανεπιθύμητων ενεργειών που εμφανίζονται μετά την έγκριση του φαρμακευτικού προϊόντος είναι σημαντική, καθώς επιτρέπει τη συνεχή παρακολούθηση της σχέσης οφέλους / κινδύνου του φαρμακευτικού προϊόντος. Οι επαγγελματίες υγείας καλούνται να αναφέρουν τυχόν υποψίες ανεπιθύμητων ενεργειών μέσω του Ιταλικού Οργανισμού Φαρμάκων. , ιστοσελίδα: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04,9 Υπερδοσολογία
Έχουν αναφερθεί περιπτώσεις τυχαίας υπερδοσολογίας από κλινικές δοκιμές σε υγρά AMD και δεδομένα μετά την κυκλοφορία. Οι ανεπιθύμητες ενέργειες που συσχετίζονται συχνότερα με αυτές τις περιπτώσεις ήταν αυξημένη ενδοφθάλμια πίεση, παροδική τύφλωση, μειωμένη οπτική οξύτητα, οίδημα του κερατοειδούς και πόνος. Εάν εμφανιστεί υπερδοσολογία, η ενδοφθάλμια πίεση πρέπει να παρακολουθείται και να αντιμετωπίζεται όπως κρίνεται απαραίτητο από το γιατρό.
05.0 ΦΑΡΜΑΚΟΛΟΓΙΚΕΣ ΙΔΙΟΤΗΤΕΣ
05.1 Φαρμακοδυναμικές ιδιότητες
Φαρμακοθεραπευτική ομάδα: Οφθαλμολογικά, αντι-νεοαγγειακά μέσα, κωδικός ATC: S01LA04
Το Ranibizumab είναι ένα εξανθρωπισμένο θραύσμα ανασυνδυασμένου μονοκλωνικού αντισώματος που κατευθύνεται κατά του ανθρώπινου αγγειακού ενδοθηλιακού αυξητικού παράγοντα Α (VEGF-A). Συνδέεται με μεγάλη συγγένεια με τις ισομορφές VEGF-A (π.χ. VEGF110, VEGF121 και VEGF165), εμποδίζοντας έτσι τη σύνδεση του VEGF-A με τους υποδοχείς VEGFR-1 και VEGFR-2. Στους υποδοχείς του οδηγεί σε πολλαπλασιασμό των ενδοθηλιακών κυττάρων σε μια νεοαγγείωση και μια αύξηση της αγγειακής διαπερατότητας, τα οποία πιστεύεται ότι συμβάλλουν στην εξέλιξη της νεοαγγειακής μορφής εκφυλισμού της ωχράς κηλίδας που σχετίζεται με την ηλικία, της παθολογικής μυωπίας ή της μειωμένης όρασης που προκαλείται είτε από διαβητικό οίδημα της ωχράς κηλίδας είτε από «οίδημα της ωχράς κηλίδας δευτερογενή μετά από RVO.
Θεραπεία υγρού AMD
Για υγρή AMD, η ασφάλεια και η κλινική αποτελεσματικότητα του Lucentis αξιολογήθηκαν σε τρεις τυχαιοποιημένες, διπλά τυφλές, εικονικές ή ενεργά ελεγχόμενες μελέτες 24 μηνών σε ασθενείς με νεοαγγειακή AMD. Συνολικά 1.323 ασθενείς (879 υπό θεραπεία και 444 μάρτυρες) εγγράφηκαν σε αυτές τις μελέτες.
Στη μελέτη FVF2598g (MARINA), 716 ασθενείς με ελάχιστα κλασικές ή απόκρυφες βλάβες χοριοειδούς νεοαγγείωσης (CNV) χωρίς κλασικό συστατικό έλαβαν μηνιαίες ενδοϋαλοειδείς ενέσεις Lucentis 0,3 mg (n = 238) ή 0,5 mg (n = 240) ή ψευδείς ενέσεις (n = 238).
Στη μελέτη FVF2587g (ANCHOR), 423 ασθενείς με κυρίως κλασικό CNV έλαβαν μία από τις ακόλουθες θεραπείες: 1) μηνιαίες ενδοϋαλοειδείς ενέσεις Lucentis 0,3 mg και PDT sham (n = 140). 2) μηνιαίες ενδοϋαλοειδείς ενέσεις Lucentis 0,5 mg και PDT sham (n = 140). ή 3) ενδοϋαλοειδείς εικονικές ενέσεις και PDT με βερτεπορφίνη (n = 143). Το PDT με βερτεπορφίνη ή ψεύτικο χορηγήθηκε μαζί με την αρχική ένεση του Lucentis και στη συνέχεια κάθε 3 μήνες εάν η φθορογγειογραφία έδειξε επιμονή ή συνέχιση της διαρροής των αγγείων.
Τα βασικά ευρήματα συνοψίζονται στους Πίνακες 1, 2 και στο Σχήμα 1.
Πίνακας 1 Αποτελέσματα τον μήνα 12 και μήνα 24 στη μελέτη FVF2598g (MARINA)
απ
Πίνακας 2 Αποτελέσματα τον μήνα 12 και μήνα 24 στη μελέτη FVF2587g (ANCHOR)
Τραπέζι
Τα αποτελέσματα και των δύο μελετών έδειξαν ότι η συνέχιση της θεραπείας με ρανιμπιζουμάμπη θα μπορούσε επίσης να είναι επωφελής σε ασθενείς που έχασαν ≥15 γράμματα καλύτερης διορθωμένης οπτικής οξύτητας (BCVA) τον πρώτο χρόνο θεραπείας.
Η μελέτη FVF3192g (PIER) ήταν μια τυχαιοποιημένη, διπλά τυφλή, ελεγχόμενη με ψεύτικη μελέτη σχεδιασμένη να αξιολογεί την ασφάλεια και την αποτελεσματικότητα του Lucentis σε 184 ασθενείς με όλες τις μορφές νεοαγγειακής AMD. Οι ασθενείς έλαβαν ενδοϋαλοειδείς ενέσεις Lucentis 0,3 mg (n = 60) ή 0,5 mg (n = 61) ή εικονικές ενέσεις (n = 63) μία φορά το μήνα για 3 συνεχόμενες δόσεις, ακολουθούμενη από μία δόση μία φορά κάθε 3 μήνες. Από τον 14ο μήνα της μελέτης, ασθενείς που έλαβαν ψευδή ένεση εισήχθησαν θεραπεία με ρανιμπιζουμάμπη και από τον μήνα 19, θα μπορούσαν να γίνουν πιο συχνές θεραπείες. Οι ασθενείς που έλαβαν θεραπεία με Lucentis στη μελέτη PIER έλαβαν κατά μέσο όρο 10 θεραπείες συνολικά.
Το κύριο τελικό σημείο αποτελεσματικότητας ήταν η μέση αλλαγή στην οπτική οξύτητα στους 12 μήνες σε σύγκριση με την αρχική τιμή. Μετά από μια αρχική αύξηση της οπτικής οξύτητας (μετά από μηνιαία δόση), κατά μέσο όρο, η οπτική οξύτητα των ασθενών μειώθηκε με την τριμηνιαία δοσολογία, επιστρέφοντας στην αρχική τιμή τον μήνα 12 και αυτό το αποτέλεσμα διατηρήθηκε στην πλειοψηφία των ασθενών που έλαβαν θεραπεία. Με ρανιμπιζουμάμπη (82%) σε Μήνας 24. Δεδομένα από περιορισμένο αριθμό ατόμων που είχαν μεταφερθεί στη θεραπεία με ρανιμπιζουμάμπη μετά από περισσότερο από ένα χρόνο ψεύτικης θεραπείας έδειξαν ότι η έγκαιρη έναρξη της θεραπείας μπορεί να σχετίζεται με καλύτερη διατήρηση της «οπτικής οξύτητας».
Και στις δύο μελέτες MARINA και ANCHOR, η βελτίωση της οπτικής οξύτητας που παρατηρήθηκε με το Lucentis 0,5 mg στους 12 μήνες συνοδεύτηκε από οφέλη που αναφέρθηκαν από τον ασθενή, όπως μετρήθηκε από τη βαθμολογία του National Eye Institute Visual Function Questionnaire (VFQ-25). Διαφορές μεταξύ Lucentis 0,5 mg και οι δύο ομάδες ελέγχου αξιολογήθηκαν με τιμές ρ που κυμαίνονται από 0,009 έως
Η αποτελεσματικότητα του Lucentis στη θεραπεία της υγρής AMD επιβεβαιώθηκε σε μελέτες AMD μετά την κυκλοφορία. Τα δεδομένα από δύο μελέτες (MONT BLANC, BPD952A2308 και DENALI, BPD952A2309) δεν κατέδειξαν επιπρόσθετα αποτελέσματα. Της συνδυασμένης χορήγησης verteporfin (Visudyne PDT) και Lucentis σε σύγκριση με τον Lucentis μόνο.
Θεραπεία της οπτικής δυσλειτουργίας λόγω DME
Η ασφάλεια και η αποτελεσματικότητα του Lucentis αξιολογήθηκαν σε δύο τυχαιοποιημένες, διπλά τυφλές, ελεγχόμενες με ψεύτικο ή ενεργές μελέτες 12 μηνών σε ασθενείς με μειωμένη όραση λόγω διαβητικού οιδήματος της ωχράς κηλίδας. Συνολικά αυτές οι μελέτες συμμετείχαν. 496 ασθενείς (336 ενεργές και 160 μάρτυρες), οι περισσότεροι είχαν διαβήτη τύπου II, 28 ασθενείς που έλαβαν θεραπεία είχαν διαβήτη τύπου Ι.
Στη φάση II της μελέτης D2201 (RESOLVE), 151 ασθενείς έλαβαν θεραπεία με ρανιμπιζουμάμπη (6 mg / ml, n = 51, 10 mg / ml, n = 51) ή ψευδή (n = 49) με μία "ενδοϋαλοειδή ένεση ανά μήνα. έως ότου τηρηθούν τα προκαθορισμένα κριτήρια. Η αρχική δόση ρανιμπιζουμάμπης (0,3 mg ή 0,5 mg) θα μπορούσε να διπλασιαστεί ανά πάσα στιγμή κατά τη διάρκεια της μελέτης μετά την πρώτη ένεση. Η φωτοπηξία με λέιζερ επιτρέπεται ως θεραπεία διάσωσης από τον 3ο μήνα και στα δύο σκέλη θεραπείας. είχε δύο μέρη: ένα εξερευνητικό μέρος (οι πρώτοι 42 ασθενείς επισκέφθηκαν τον μήνα 6) και ένα επιβεβαιωτικό μέρος (οι υπόλοιποι 109 ασθενείς επισκέφθηκαν τον μήνα 12).
Τα βασικά ευρήματα από το επιβεβαιωτικό μέρος της μελέτης (τα 2/3 των ασθενών) συνοψίζονται στον Πίνακα 3.
Πίνακας 3 Αποτελέσματα τον μήνα 12 στη Μελέτη D2201 (ΑΠΟΦΑΣΗ) (Συνολικός πληθυσμός μελέτης)
απ
Στη μελέτη φάσης III D2301 (RESTORE), 345 ασθενείς με προβλήματα όρασης λόγω οιδήματος της ωχράς κηλίδας τυχαιοποιήθηκαν για να λάβουν είτε «ενδοϋαλοειδή ένεση 0,5 mg ρανιμπιζουμάμπης ως μονοθεραπεία και φωτοπηξία με λέιζερ (n = 116), είτε συνδυασμό 0,5 mg ρανιμπιζουμάμπη και φωτοπηξία με λέιζερ (n = 118) ή "ψευδή ένεση και φωτοπηξία με λέιζερ (n = 111). Η θεραπεία με ρανιμπιζουμάμπη ξεκίνησε με μηνιαίες ενδοϋαλοειδείς ενέσεις και συνεχίστηκε έως ότου η οπτική οξύτητα παρέμεινε σταθερή για τουλάχιστον τρεις διαδοχικούς μηνιαίους ελέγχους. Η θεραπεία συνεχίστηκε όταν παρατηρήθηκε μείωση του BCVA λόγω της εξέλιξης του DME. Η φωτοπηξία με λέιζερ χορηγήθηκε κατά την έναρξη την ίδια ημέρα, τουλάχιστον 30 λεπτά πριν από την ένεση ρανιμπιζουμάμπης και στη συνέχεια, όπως απαιτείται, βάσει κριτηρίων ETDRS.
Τα βασικά ευρήματα συνοψίζονται στον Πίνακα 4 και στο Σχήμα 2.
Πίνακας 4 Αποτελέσματα τον Μήνα 12 στη Μελέτη D2301 (ΑΠΟΚΑΤΑΣΤΑΣΗ)
απ
Το αποτέλεσμα ήταν συνεπές στις περισσότερες υποομάδες. Ωστόσο, άτομα με αρκετά υψηλό BCVA στην αρχή (> 73 γράμματα) με οίδημα της ωχράς κηλίδας και κεντρικό πάχος αμφιβληστροειδούς
Η βελτίωση της οπτικής οξύτητας τον Μήνα 12 που παρατηρήθηκε με το Lucentis 0,5 mg συνοδεύτηκε από οφέλη που αναφέρθηκαν από ασθενείς από σημαντικές λειτουργίες που σχετίζονται με την όραση, όπως μετρήθηκε από το σκορ του National Eye Institute Visual Function Questionnaire (VFQ-25). Δεν θα μπορούσαν να υπάρξουν διαφορές λόγω θεραπείας Η διαφορά μεταξύ Lucentis 0,5 mg και της ομάδας ελέγχου αξιολογήθηκε με τιμή p 0,0137 (ranibizumab mono) και 0,0041 (ranibizumab + laser) για τη σύνθετη βαθμολογία του VFQ-25.
Και στις δύο μελέτες, η οπτική βελτίωση συνοδεύτηκε από μια συνεχή μείωση του οιδήματος της ωχράς κηλίδας που μετρήθηκε ως κεντρικό πάχος αμφιβληστροειδούς (CRT).
Θεραπεία της οπτικής δυσλειτουργίας που προκαλείται από οίδημα της ωχράς κηλίδας δευτερογενή σε RVO
Η κλινική ασφάλεια και αποτελεσματικότητα του Lucentis σε ασθενείς με προβλήματα όρασης λόγω οίδημα της ωχράς κηλίδας που οφείλεται σε RVO αξιολογήθηκαν σε τυχαιοποιημένες, διπλά τυφλές, ελεγχόμενες δοκιμές: BRAVO και CRUISE που στρατολόγησαν ασθενείς με BRVO (n = 397) και CRVO (n = 392 Και στις δύο μελέτες, οι ασθενείς έλαβαν ενδοϋαλοειδείς ή ψευδείς ενέσεις ρανιβιζουμάμπης 0,3 mg ή 0,5 mg. Μετά από 6 μήνες, οι ασθενείς στον βραχίονα ελέγχου μετακινήθηκαν στην ομάδα ρανιμπιζουμάμπης 0,5 mg. Στη μελέτη BRAVO, η φωτοπηξία με λέιζερ ως θεραπεία διάσωσης επιτρέπεται σε όλα τα χέρια από τον 3ο μήνα.
Βασικά ευρήματα από τις μελέτες BRAVO και CRUISE παρουσιάζονται στους Πίνακες 5 και 6
Πίνακας 5 Αποτελέσματα τον μήνα 6 και 12 (BRAVO)
απ
Πίνακας 6 Αποτελέσματα τον μήνα 6 και 12 (CRUISE)
απ
Και στις δύο μελέτες, η οπτική βελτίωση συνοδεύτηκε από μια συνεχή και σημαντική μείωση του οιδήματος της ωχράς κηλίδας που μετρήθηκε ως προς το πάχος του κεντρικού αμφιβληστροειδούς.
Σε ασθενείς με BRVO (μελέτη BRAVO και επέκταση μελέτης HORIZON): Μετά από 2 χρόνια, οι ασθενείς που είχαν λάβει θεραπεία με ψεύτικες ενέσεις τους πρώτους 6 μήνες και στη συνέχεια μεταφέρθηκαν σε θεραπεία με ranibizumab είχαν κέρδος AV (& symp; 15 γράμματα) συγκρίσιμο με αυτό ασθενών που είχαν λάβει θεραπεία με ρανιμπιζουμάμπη από την έναρξη της μελέτης (& symp; 16 γράμματα). Ωστόσο, ο αριθμός των ασθενών που συμπλήρωσαν 2 χρόνια ήταν περιορισμένος και προγραμματίστηκαν μόνο τριμηνιαίες επισκέψεις στη μελέτη HORIZON. υπάρχουν επαρκή στοιχεία για να καταλήξουμε με συστάσεις για πότε πρέπει να ξεκινά η θεραπεία με ranibizumab σε ασθενείς με BRVO.
Σε ασθενείς με CRVO (μελέτη CRUISE και επέκταση της μελέτης HORIZON): Μετά από 2 χρόνια, οι ασθενείς που είχαν λάβει θεραπεία τους πρώτους 6 μήνες με ψεύτικες ενέσεις και στη συνέχεια μεταφέρθηκαν σε θεραπεία με ranibizumab δεν έδειξαν κέρδη στην AV (& symp; 6 γράμματα) σε σύγκριση με εκείνους ασθενών που είχαν λάβει θεραπεία με ranibizumab από την έναρξη της μελέτης (& symp; 12 γράμματα).
Η βελτίωση της οπτικής οξύτητας που παρατηρήθηκε με τη θεραπεία με ρανιμπιζουμάμπη στους μήνες 6 και 12 συνοδεύτηκε από οφέλη που αναφέρθηκαν από τον ασθενή, όπως μετρήθηκαν από την υποομάδα του Εθνικού Ινστιτούτου Οφθαλμικής Οπτικής Λειτουργίας (NEI VFQ-25) κοντινών και μακρινών δραστηριοτήτων. Η διαφορά μεταξύ Lucentis 0,5 mg και η ομάδα ελέγχου βρέθηκε να είναι μεταξύ των τιμών ρ μεταξύ 0,02 και 0,0002.
Θεραπεία της οπτικής δυσλειτουργίας λόγω CNV δευτεροπαθούς σε PM
Η ασφάλεια και η κλινική αποτελεσματικότητα του Lucentis σε ασθενείς με προβλήματα όρασης λόγω CNV σε PM επικυρώθηκαν με βάση δεδομένα δώδεκα μηνών από την κεντρική τυχαιοποιημένη, διπλά τυφλή, ελεγχόμενη μελέτη F2301 (RADIANCE). Αυτή η μελέτη είχε ως στόχο να αξιολογήσει δύο διαφορετικά δοσολογικά σχήματα της ρανιμπιζουμάμπης 0,5 mg που χορηγήθηκε με ενδοϋαλοειδή ένεση έναντι της βερτεπορφίνης PDT (vPDT, φωτοδυναμική θεραπεία Visudyne). Οι 277 ασθενείς τυχαιοποιήθηκαν σε έναν από τους ακόλουθους βραχίονες:
• Ομάδα Ι (ρανιμπιζουμάμπη 0,5 mg, θεραπευτικό σχήμα που καθορίζεται από κριτήρια "σταθερότητας" που ορίζονται ως καμία αλλαγή στην BCVA σε σύγκριση με τις εκτιμήσεις των δύο προηγούμενων μηνών).
• Ομάδα II (ρανιμπιζουμάμπη 0,5 mg, θεραπευτικό σχήμα που καθορίζεται από κριτήρια "δραστηριότητας της νόσου" που ορίζονται ως διαταραχή της όρασης που οφείλεται σε ενδο- ή υπο-αμφιβληστροειδές υγρό ή ενεργή διαρροή που προκαλείται από βλάβες CNV, όπως αποδεικνύεται από OCT και / ή AF).
• Ομάδα III (ασθενείς που λαμβάνουν θεραπεία με vPDT - με δυνατότητα θεραπείας με ranibizumab από τον 3ο μήνα).
Κατά τη διάρκεια των 12 μηνών της μελέτης, οι ασθενείς έλαβαν κατά μέσο όρο 4,6 ενέσεις (εύρος 1-11) στην ομάδα Ι και 3,5 ενέσεις (εύρος 1-12) στην ομάδα II. Μεταξύ των ασθενών που ανήκουν στην Ομάδα II, η οποία αντικατοπτρίζει τη συνιστώμενη δοσολογία (βλέπε παράγραφο 4.2), το 50,9% των ασθενών υποβλήθηκαν σε θεραπεία με 1 έως 2 ενέσεις, 34,5% 3 έως 5 ενέσεις και 14,7% έδωσαν 6 έως 12 ενέσεις κατά τη διάρκεια της 12μηνης μελέτης Το Το 62,9% των ασθενών της ομάδας ΙΙ δεν χρειάστηκε καμία ένεση κατά τους δεύτερους 6 μήνες της μελέτης.
Τα βασικά ευρήματα από το RADIANCE συνοψίζονται στον Πίνακα 7 και στο Σχήμα 5.
Πίνακας 7 Αποτελέσματα στον μήνα 3 και 12 (ΑΚΤΙΝΟΒΟΛΙΑ)
απ
β Συγκριτικός έλεγχος έως τον μήνα 3. Οι ασθενείς που τυχαιοποιήθηκαν να λάβουν vPDT ήταν επιλέξιμοι για θεραπεία με ranibizumab τον μήνα 3 (στην ομάδα III, 38 ασθενείς έλαβαν ranibizumab τον μήνα 3)
Η βελτίωση της όρασης συνοδεύτηκε από μείωση του πάχους του κεντρικού αμφιβληστροειδούς.
Σε σύγκριση με την ομάδα που έλαβε θεραπεία με vPDT, οι ασθενείς στις ομάδες που έλαβαν ranibizumab ανέφεραν όφελος (τιμή p
Παιδιατρικός πληθυσμός
Η ασφάλεια και η αποτελεσματικότητα του ranibizumab σε παιδιά δεν έχουν ακόμη τεκμηριωθεί.
Ο Ευρωπαϊκός Οργανισμός Φαρμάκων παραιτήθηκε από την υποχρέωση υποβολής των αποτελεσμάτων των μελετών με το Lucentis σε όλα τα υποσύνολα του παιδιατρικού πληθυσμού για νεοαγγειακή AMD, διαταραχή της όρασης λόγω DME, οπτική δυσλειτουργία λόγω δευτερογενούς οιδήματος της ωχράς κηλίδας σε RVO και οπτική δυσλειτουργία λόγω CNV δευτεροπαθούς PM (βλ. Παράγραφο 4.2 για πληροφορίες σχετικά με την παιδιατρική χρήση).
05.2 Φαρμακοκινητικές ιδιότητες
Μετά από μηνιαία ενδοϋαλοειδή χορήγηση του Lucentis σε ασθενείς με νεοαγγειακή AMD, οι συγκεντρώσεις του ranibizumab στον ορό ήταν γενικά χαμηλές, με τα μέγιστα επίπεδα (Cmax) γενικά κάτω από τη συγκέντρωση ρανιμπιζουμάμπης που απαιτείται για την αναστολή της βιολογικής δραστηριότητας του VEGF κατά 50% (11-27 ng / mL , αξιολογήθηκε σε δοκιμή in vitro κυτταρικός πολλαπλασιασμός). Η Cmax ήταν ανάλογη με τη δόση σε όλο το εύρος δόσεων από 0,05 έως 1,0 mg / μάτι. Σε περιορισμένο αριθμό ασθενών με DME, οι ανιχνευμένες συγκεντρώσεις στον ορό δείχνουν ότι δεν μπορεί να αποκλειστεί μια ελαφρώς υψηλότερη συστηματική έκθεση σε αυτούς που παρατηρήθηκαν σε ασθενείς με νεοαγγειακή AMD. Οι συγκεντρώσεις του ranibizumab στον ορό σε ασθενείς με RVO ήταν παρόμοιες ή ελαφρώς υψηλότερες από αυτές που παρατηρήθηκαν σε ασθενείς με νεοαγγειακή AMD.
Με βάση τη φαρμακοκινητική ανάλυση πληθυσμού και την κάθαρση του ranibizumab στον ορό για νεοαγγειακούς ασθενείς με AMD που έλαβαν δόση 0,5 mg, ο μέσος χρόνος ημίσειας ζωής αποβολής του ranibizumab από υαλοειδή είναι περίπου 9 ημέρες. Κατά τη μηνιαία ενδοϋαλοειδή χορήγηση του Lucentis 0,5 mg / μάτι, ο ορός C του ranibizumab, που έφτασε περίπου 1 ημέρα μετά τη δόση, αναμένεται να κυμαίνεται γενικά μεταξύ 0,79 και 2,90 ng / ml, ενώ αναμένεται ότι το Cmin κυμαίνεται γενικά μεταξύ 0,07 και 0,49 ng / ml. Οι συγκεντρώσεις του ranibizumab στον ορό εκτιμάται ότι είναι περίπου 90.000 φορές χαμηλότερες από τις συγκεντρώσεις υαλοειδούς.
Ασθενείς με νεφρική ανεπάρκεια: Δεν έχουν πραγματοποιηθεί συμβατικές μελέτες για την εξέταση της φαρμακοκινητικής του Lucentis σε ασθενείς με νεφρική ανεπάρκεια. Σε μια "φαρμακοκινητική ανάλυση σε πληθυσμό νεοαγγειακών ασθενών με AMD, το 68% (136 στους 200) των ασθενών είχαν" νεφρική ανεπάρκεια (46,5% ήπια [50-80 mL / min], 20% μέτρια [30 -50 mL / min] και 15% σοβαρή [η συστηματική κάθαρση ήταν ελαφρώς χαμηλότερη, αλλά αυτό δεν ήταν κλινικά σημαντικό.
Ασθενείς με ηπατική ανεπάρκεια: Δεν έχουν πραγματοποιηθεί συμβατικές μελέτες για την εξέταση της φαρμακοκινητικής του Lucentis σε ασθενείς με ηπατική ανεπάρκεια.
05.3 Προκλινικά δεδομένα ασφάλειας
Η διμερής ενδοϋαλοειδής χορήγηση ρανιμπιζουμάμπης σε πιθήκους cynomolgus σε δόσεις μεταξύ 0,25 mg / μάτι και 2,0 mg / μάτι μία φορά κάθε 2 εβδομάδες για έως και 26 εβδομάδες είχε ως αποτέλεσμα τη δοσοεξαρτώμενη οφθαλμική δράση.
Ενδοφθάλμια, εξαρτώμενες από τη δόση αυξήσεις της έξαρσης και των κυττάρων σημειώθηκαν στον πρόσθιο θάλαμο, κορυφώνοντας 2 ημέρες μετά την ένεση. Η σοβαρότητα της φλεγμονώδους απόκρισης μειώνεται γενικά με τις επόμενες ενέσεις ή κατά τη διάρκεια της περιόδου ανάρρωσης. Στο οπίσθιο τμήμα εμφανίστηκαν κυτταρικές διηθήσεις και υαλοειδείς πλωτήρες η οποία έτεινε επίσης να εξαρτάται από τη δόση και γενικά παρέμεινε μέχρι το τέλος της περιόδου θεραπείας.Στη μελέτη 26 εβδομάδων, η σοβαρότητα της φλεγμονής του υαλοειδούς αυξήθηκε με τον αριθμό των ενέσεων. Ωστόσο, παρατηρήθηκε αναστρεψιμότητα μετά την περίοδο αποκατάστασης. Η φύση και η διάρκεια της φλεγμονής του οπίσθιου τμήματος είναι ενδεικτική μιας ανοσολογικής απόκρισης αντισωμάτων, η οποία μπορεί να είναι κλινικά άσχετη. Ο σχηματισμός καταρράκτη έχει παρατηρηθεί σε ορισμένα ζώα μετά από μια σχετικά μεγάλη περίοδο έντονης φλεγμονής, υποδηλώνοντας ότι οι αλλαγές του φακού ήταν δευτερεύουσες σε σοβαρή φλεγμονή. Παρατηρήθηκε παροδική αύξηση της ενδοφθάλμιας πίεσης μετά τη χορήγηση, ανεξάρτητα από τη δόση, μετά από ενδοϋαλοειδείς ενέσεις.
Οι μικροσκοπικές οφθαλμικές αλλαγές σχετίζονται με φλεγμονή και δεν υποδηλώνουν εκφυλιστικές διεργασίες. Φλεγμονώδεις κοκκιωματώδεις αλλαγές σημειώθηκαν στον οπτικό δίσκο μερικών ματιών. Αυτές οι μεταγενέστερες αλλαγές τμήματος μειώθηκαν και σε ορισμένες περιπτώσεις επιλύθηκαν, κατά την περίοδο της ανάρρωσης.
Δεν υπήρχαν σημάδια συστηματικής τοξικότητας μετά από ενδοϋαλοειδή χορήγηση. Αντισώματα ορού και υαλοειδούς στο ranibizumab βρέθηκαν σε ένα υποσύνολο ζώων που έλαβαν θεραπεία.
Δεν υπάρχουν διαθέσιμα στοιχεία καρκινογένεσης ή μεταλλαξιογένεσης.
Σε εγκύους πιθήκους, η ενδοϋαλώδης ένεση ρανιμπιζουμάμπης με αποτέλεσμα μέγιστη συστηματική έκθεση 0,9-7 φορές τη χειρότερη κλινική έκθεση δεν προκάλεσε αναπτυξιακή τοξικότητα ή τερατογονικότητα και δεν είχε καμία επίδραση στο βάρος ή τη δομή του σώματος. Πλακούντας, αν και το ranibizumab θα πρέπει ενδεχομένως να εξεταστεί τερατογόνο και έμβρυο / εμβρυοτοξικό με βάση τη φαρμακολογική του επίδραση.
Η απουσία μεσολαβούμενων επιδράσεων της ρανιμπιζουμάμπης στην ανάπτυξη του εμβρύου / εμβρύου σχετίζεται εύλογα κυρίως με την αδυναμία του θραύσματος Fab να διασχίσει τον πλακούντα. Ωστόσο, περιγράφηκε μια περίπτωση με υψηλά επίπεδα ranibizumab στον ορό της μητέρας και παρουσία ranibizumab στον εμβρυϊκό ορό, υποδηλώνοντας ότι το αντίσωμα κατά της ranibizumab λειτούργησε ως πρωτεΐνη (που περιέχει την περιοχή FC) που μεταφέρει τη ranibizumab, μειώνοντας έτσι την αποβολή της από τον μητρικό ορό και επιτρέποντας τη μεταφορά του στον πλακούντα. Δεδομένου ότι οι δοκιμές για την ανάπτυξη εμβρύου / εμβρύου έχουν διεξαχθεί σε υγιή έγκυα ζώα και ορισμένες ασθένειες (όπως ο διαβήτης) μπορούν να τροποποιήσουν τη διαπερατότητα του πλακούντα σε ένα κομμάτι Fab, η μελέτη πρέπει να ερμηνευτεί με προσοχή.
06.0 ΦΑΡΜΑΚΕΥΤΙΚΕΣ ΠΛΗΡΟΦΟΡΙΕΣ
06.1 Έκδοχα
διένυδρη α, α-τρεχαλόζη
Υδροχλωρική ιστιδίνη, μονοένυδρη
Ιστιδίνη
Πολυσορβικό 20
Νερό για ενέσεις
06.2 Ασυμβατότητα
Ελλείψει μελετών συμβατότητας, αυτό το φαρμακευτικό προϊόν δεν πρέπει να αναμειγνύεται με άλλα φαρμακευτικά προϊόντα.
06.3 Περίοδος ισχύος
3 χρόνια
06.4 Ειδικές προφυλάξεις κατά την αποθήκευση
Φυλάσσετε σε ψυγείο (2 ° C - 8 ° C).
Μην παγώνετε.
Φυλάξτε το φιαλίδιο στο εξωτερικό κουτί για να προστατεύσετε το φάρμακο από το φως.
06.5 Φύση της άμεσης συσκευασίας και περιεχόμενο της συσκευασίας
0,23 ml αποστειρωμένο διάλυμα σε φιαλίδιο (γυαλί τύπου Ι) με πώμα (ελαστικό χλωροβουτυλίου), 1 αμβλύ βελόνα φίλτρου (18G x 1½ ", 1,2 mm x 40 mm, 5 mcm), 1 βελόνα ένεσης (30G x ½", 0,3 mm x 13 mm) και 1 σύριγγα (πολυπροπυλένιο) (1 ml). Η συσκευασία περιέχει 1 φιαλίδιο.
06.6 Οδηγίες χρήσης και χειρισμού
Το φιαλίδιο, η βελόνα ένεσης, η βελόνα φίλτρου και η σύριγγα προορίζονται μόνο για μία χρήση. Η επαναχρησιμοποίηση μπορεί να προκαλέσει μόλυνση ή άλλη ασθένεια / τραυματισμό. Όλα τα συστατικά είναι αποστειρωμένα. Δεν πρέπει να χρησιμοποιείται οποιοδήποτε εξάρτημα με συσκευασία που δείχνει σημάδια φθοράς ή παραποίησης. Η στειρότητα δεν μπορεί να εγγυηθεί εάν η σφραγίδα συσκευασίας δεν είναι άθικτη.
Για να προετοιμάσετε το Lucentis για ενδοϋαλοειδή ένεση, ακολουθήστε τις παρακάτω οδηγίες:
1. Απολυμάνετε το εξωτερικό του ελαστικού πώματος του φιαλιδίου πριν από τη συλλογή.
2. Συνδέστε ασηπτικά τη βελόνα φίλτρου 5 mcm (18G x 1½ ", 1,2 mm x 40 mm, παρέχεται) σε μια σύριγγα 1 ml (παρέχεται). Τοποθετήστε τη αμβλύ βελόνα φίλτρου στο κέντρο του πώματος μέχρι να αγγίξει το κάτω μέρος του φιαλιδίου.
3. Αφαιρέστε όλο το υγρό από το φιαλίδιο κρατώντας το σε όρθια θέση, ελαφρώς κεκλιμένο για να διευκολύνετε την πλήρη απόσυρση.
4. Βεβαιωθείτε ότι το έμβολο της σύριγγας τραβιέται αρκετά πίσω όταν αδειάζετε το φιαλίδιο για να αδειάσει τελείως τη βελόνα φίλτρου.
5. Αφήστε τη βελόνα φίλτρου αμβλύ στο φιαλίδιο και αφαιρέστε τη σύριγγα από αυτήν. Πετάξτε τη βελόνα φίλτρου μετά την απόσυρση του περιεχομένου του φιαλιδίου και μην τη χρησιμοποιήσετε για ενδοϋαλοειδή ένεση.
6. Συνδέστε τη βελόνα ένεσης (30G x ½ ", 0.3mm x 13mm, παρέχεται) με ασφάλεια και ασηπτικά στη σύριγγα.
7. Αφαιρέστε προσεκτικά το πώμα από τη βελόνα ένεσης χωρίς να αποσυνδέσετε τη βελόνα ένεσης από τη σύριγγα.
Σημείωση: Κρατήστε την κίτρινη βάση της βελόνας ένεσης ενώ αφαιρείτε το πώμα.
8. Αφαιρέστε προσεκτικά τον αέρα από τη σύριγγα και ρυθμίστε τη δόση στα 0,05 ml που αναγράφεται στη σύριγγα.Η σύριγγα είναι έτοιμη για ένεση.
Σημείωση: Μην καθαρίζετε τη βελόνα ένεσης, μην τραβάτε το έμβολο προς τα πίσω.
Μετά την ένεση, μην καλύπτετε τη βελόνα και μην την αποσυνδέετε από τη σύριγγα. Απορρίψτε τη χρησιμοποιημένη σύριγγα μαζί με τη βελόνα σε κατάλληλο περιέκτη ή σύμφωνα με τις τοπικές απαιτήσεις.
07.0 ΚΑΤΟΧΟΣ ΑΔΕΙΑΣ ΜΑΡΚΕΤΙΝΓΚ
Novartis Europharm Limited
Wimblehurst Road
Χόρσαμ
Δυτικό Σάσεξ, RH12 5AB
Ηνωμένο Βασίλειο
08.0 ΑΡΙΘΜΟΣ ΑΔΕΙΑΣ ΜΑΡΚΕΤΙΝΓΚ
ΕΕ/1/06/374/001
037608027
09.0 ΗΜΕΡΟΜΗΝΙΑ ΠΡΩΤΗΣ ΕΓΚΡΙΣΗΣ OR ΑΝΑΝΕΩΣΗΣ ΤΗΣ ΑΔΕΙΑΣ
Ημερομηνία πρώτης έγκρισης: 22 Ιανουαρίου 2007
Ημερομηνία πρόσφατης ανανέωσης: 24 Ιανουαρίου 2012
10.0 ΗΜΕΡΟΜΗΝΙΑ ΑΝΑΘΕΩΡΗΣΗΣ ΤΟΥ ΚΕΙΜΕΝΟΥ
05/2014